
 |
By Jamshed Arslan Pharm.D.
The cell’s oxygen-sensing machinery comprises prolyl-4-hydroxylases (P4Hs 1-3, PHDs 1-3, or EGLN 1-3) and their canonical target hypoxia-inducible factors (HIFs). When oxygen levels are low, PHDs become functionally inactive, leading to HIFs’ stability. PHD1 is thought to provide a link between hypoxia and inflammation, partly because it influences the prototypical proinflammatory transcription factor NF-kB in such a way that suppressing PHD1 reduces inflammation. However, the exact targets of PHD1 in NF-kB signaling are unclear. Moreover, PHD1 inhibition also prevents the activation of tumor suppressor p53 following chemotherapy in colorectal cancer cells. So, the researchers at University of Oulu, Finland, set out to explore the potential of PHD1 as a common regulator of hypoxia, inflammation, and cancer. They found that PHD1 inhibition downregulates proinflammatory genes and upregulates proapoptotic genes via negative regulation of NF-kB and p53 signaling.
The initial clue of the relationship between PHD1, inflammation and apoptosis came from microarray analysis of mouse embryonic fibroblasts (MEFs). Of the 1,093 differentially-expressed genes in MEFs from PHD1-null mouse line, 2.7% and 5.7% were involved in regulating inflammation and apoptosis, respectively. Quantitative RT-PCR confirmed a decrease in the mRNA levels of inflammation-related genes (Fcgr3, C1qa, C1qc, Cxcl10), but an increase in the apoptosis-related genes (Perp, Bnip3).
Likewise, mRNA levels of proinflammatory cytokines (Il6, Cxcl10, Tnfalpha) and enzymes (Cox2) were reduced in the presence of inflammatory stimulus (LPS) in the PHD1-knockout MEFs. At baseline and with LPS-stimulus, PHD1-knockout MEFs showed a decrease in both NF-kB complex proteins (Rel-A/p65, p150, p50) and NF-kB reporter activity. To see whether these effects were dependent on HIF1alpha, PHD1 was silenced (chemically and with siRNA) in HIF1alpha-double-knockout cells. Silencing PHD1 reduced NF-kB luciferase activity, indicating that PHD1 directly targets NF-kB signaling.
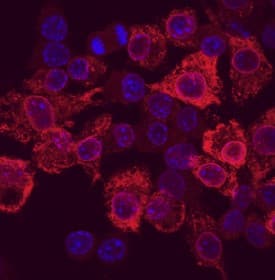 COX-2 was detected in immersion fixed RAW 264.7 mouse monocyte/macrophage cells stimulated with LPS using Goat Anti-Human/Mouse COX-2 Antigen Affinity-purified Polyclonal Antibody (Catalog # AF4198) at 10 µg/mL for 3 hours at room temperature. Cells were stained using the NorthernLights™ 557-conjugated Anti-Goat IgG Secondary Antibody (red; Catalog # NL001) and counterstained with DAPI (blue). Specific staining was localized to cytoplasm. View our protocol for Fluorescent ICC Staining of Cells on Coverslips.
COX-2 was detected in immersion fixed RAW 264.7 mouse monocyte/macrophage cells stimulated with LPS using Goat Anti-Human/Mouse COX-2 Antigen Affinity-purified Polyclonal Antibody (Catalog # AF4198) at 10 µg/mL for 3 hours at room temperature. Cells were stained using the NorthernLights™ 557-conjugated Anti-Goat IgG Secondary Antibody (red; Catalog # NL001) and counterstained with DAPI (blue). Specific staining was localized to cytoplasm. View our protocol for Fluorescent ICC Staining of Cells on Coverslips.
PHD1-knockout MEFs also showed a differential increase in p53 protein levels but not mRNA in normoxia. The increase in p53 half-life in PHD1-knockout MEFs following treatment with a protein-synthesis-inhibitor (cycloheximide), and reduced ubiquitination of p53 by silencing PHD1, confirmed that PHD1 affects p53 at a post-transcriptional level. To explore this mechanism further, overexpressed Flag-tagged p53, from HCT-116 cells treated with an anticancer compound (cisplatin) as well as a proteasomal-inhibitor, was immunoprecipitated and subjected to LC-MS analysis. Data on several p53 variants revealed that the hydroxylation target of PHD1 is Proline142. The involvement of HIF-1alpha in this process was evident from the observation that in HIF1alpha- knockouts, V5-epitope-tagged PHD1 did not coimmunoprecipitate with p53, and hydroxylation of p53 was absent.
The researchers applied proinflammatory 12-O-tetradecanoylphorbol-13-acetate (TPA) to the skin of wild-type and PHD1-knockout mice. As expected, the number of intradermal inflammatory cells and mRNA levels of Tnfalpha and Il6 were lower in the TPA-treated knockouts. In line with the in vitro data, non-treated and TPA-treated skin cells in the PHD1-knockouts showed a greater number of cells undergoing apoptosis.
This study provides a single solution, PHD1 inhibition, for two of the biggest problems of modern medicine: inflammation and cancer. The choice of multiple cell lines, the selection of prototypical pathways of inflammation (NF-kB signaling) and cancer (p53 signaling), and the validation of results in vivo enhance the chances of reproducibility of current findings in humans.
Read New Hypoxia in Cancer White Paper
 Jamshed Arslan, Pharm D.
Jamshed Arslan, Pharm D.
University of Alabama at Birmingham, School of Medicine
Dr. Arslan studies cell signaling in mitochondrial defects in C. elegans
and transgenic mice.
References
Ullah, Karim, et al. “Hypoxia-Inducible Factor Prolyl-4-Hydroxylase-1 is a Convergent Point in the Reciprocal Negative Regulation of NF-κB and p53 Signaling Pathways.” Scientific Reports, vol. 7, 2017, n. pag. DOI:10.1038/s41598-017-17376-0
